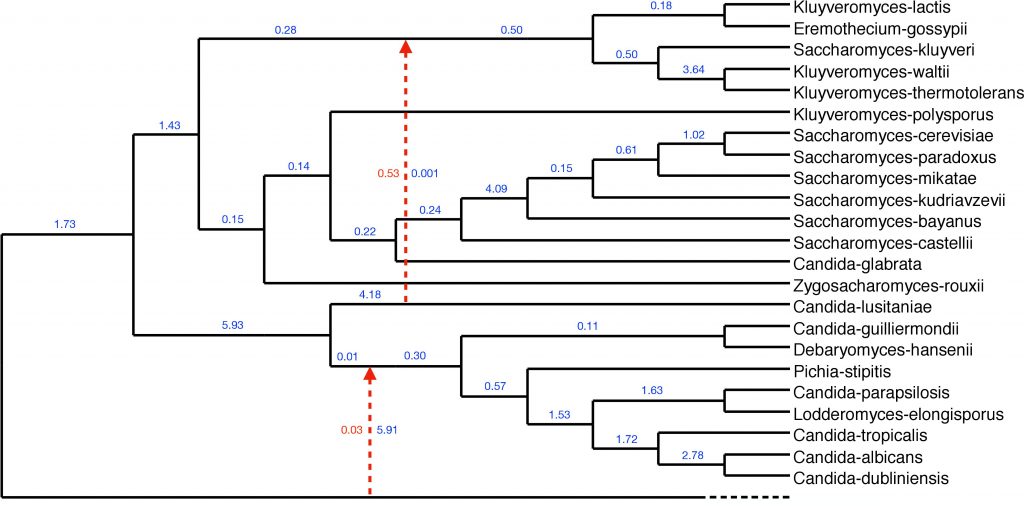
Statistical Inference of Phylogenetic Networks
The Liu and Bonito labs have created a new method for scalable statistical inference of phylogenetic networks from large-scale genomic sequence datasets. The new method has been published as part of peer-reviewed proceedings of RECOMB-CG 2018. The publication can be accessed here.
In related work, members of the Liu lab have developed a novel non-parametric/semi-parametric method for statistical support estimation. The approach has initially been applied to classical problems in computational biology and bioinformatics. The work was also published as part of peer-reviewed proceedings of RECOMB-CG 2018. The publication can be accessed here. An extended journal paper has been submitted to Algorithms for Molecular Biology.